FDA Meeting Series: How, When and What – General Introduction

Published on: Jul 21, 2022
The Food and Drug Administration (FDA) offers sponsors many opportunities for interaction and guidance during the course of drug development through routine formal meeting types including Type A, B, B (End of Phase (EOP)) and C meetings as outlined in Formal Meetings Between the FDA and Sponsors or Applicants of PDUFA Products Guidance for Industry. It is advantageous to interact early and as often as needed with the agency to ensure agreement with development and regulatory strategies and avoid time consuming and costly pitfalls.
Type A meetings are meetings necessary to resume an otherwise stalled product development program or to address important safety issues. Examples of Type A meetings include:
-
Dispute resolution meetings as described in 21 CFR 10.75, 312.48, and 314.103 and in the guidance Formal Dispute Resolution: Sponsor Appeals Above the Division Level
-
Meetings to discuss clinical holds in which development has stalled and a new path forward should be discussed
-
Special protocol assessment meetings that are requested by the sponsor after receipt of FDA evaluation of protocols under special protocol assessment procedures.
-
Post-action meetings requested within 3 months of an FDA regulatory action (besides approval)
-
Meetings requested within 30 days of an FDA issuance of a refuse-to-file letter
Type B meetings are the most prevalent meeting type and are associated with major milestones in the product development program. Type B meetings include:
-
Pre-emergency use authorization meetings
-
Pre-new drug application (pre-NDA)/pre-biologics license application (pre-BLA) meetings
-
Recently, FDA has implemented Type B meetings to review the integrated summary of efficacy (ISE) and integrated summary of safety (ISS). This meeting is meant to assist sponsors on preparation of the ISS & ISE and is scheduled after the prefiling meeting and well before submission of the marketing application
-
Post-action meetings requested 3 or more months after an FDA regulatory action (besides approval)
-
Meetings regarding risk evaluation and mitigation strategies or post marketing requirements that occur outside the context of the review of a marketing application
-
Meetings held to discuss the overall development program for products granted breakthrough therapy designation status
Type B (EOP) meetings are meetings that occur when development has reached the end of a particular phase and is ready to move to the next. Type B (EOP) meetings include:
-
End-of-phase 1 (EOP1) meetings for biologics that treat life-threatening and severely debilitating illnesses (21 CFR 312 subpart E) and drugs under accelerated approval for serious/life threatening illnesses (21 CFR 314 subpart H)
Type C meetings are meetings that cover any topic not otherwise covered by a Type A, Type B, or Type B (EOP) meeting regarding the development and review of a product. Examples include:
-
OTC Monograph Feedback (560FB) meetings
-
Phase 4 (PF_4) meetings
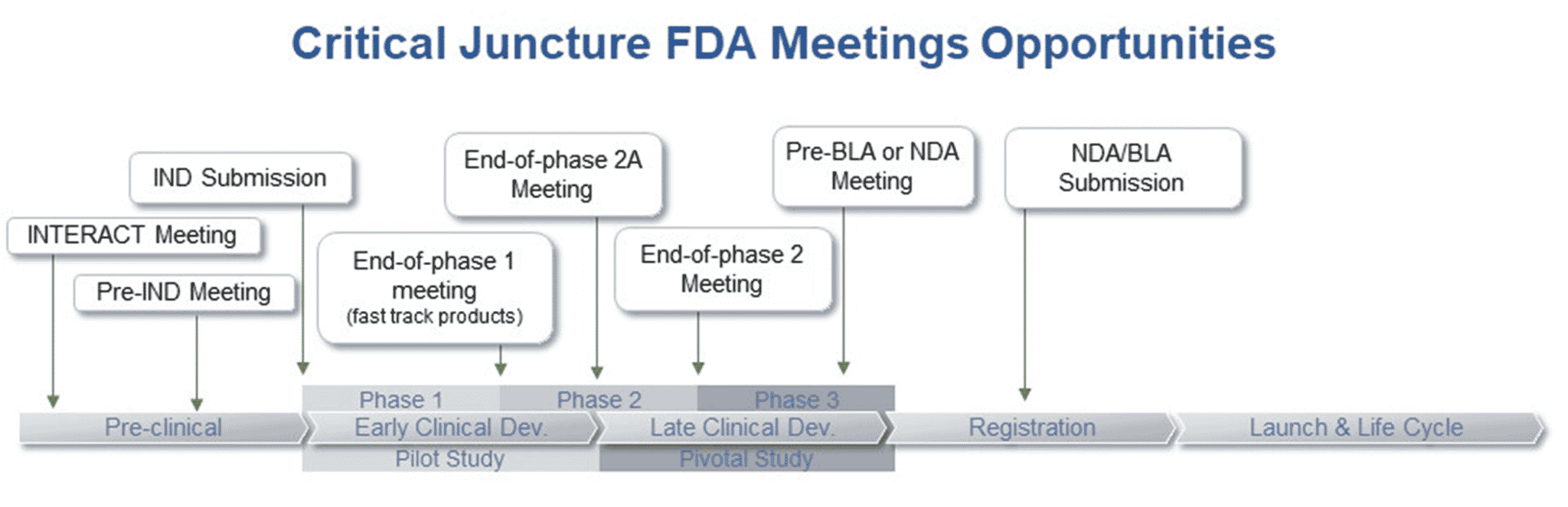
Although there are many types of meetings that can occur throughout the drug development process there are a few that occur during critical junctures. These meetings are INTERACT, pre-IND, EOP1, EOP2A, EOP2, and Pre-NDA/Pre-BLA meetings. A brief description of each has been provided below.
INTERACT Meetings
INitial Targeted Engagement for Regulatory Advice on CBER products (INTERACT) meetings are informal meetings held between sponsors of innovative investigational biological products and the Center for Biological Evaluation and Research (CBER) to obtain non-binding advice from FDA regarding chemistry, manufacturing, and controls (CMC), pharmacology/toxicology, and/or clinical aspects of the development program. CBER recognizes that the development of innovative investigational products can introduce unique challenges related to unknown safety profiles, complex manufacturing processes, technologies and issues, incorporation of innovative devices and the use of cutting-edge testing methodologies that can benefit from early agency input. The Center for Drug Evaluation and Research (CDER) does not currently have a similar meeting, but this option will become available in fiscal year 2023 when PDUFA VII is reauthorized. Upon reauthorization all INTERACT meetings will become formal PDUFA meetings.
Pre-IND Meetings
Pre-Investigational New Drug application (Pre-IND) meetings are formal PDUFA Type B meetings held between a sponsor and CDER or CBER. Prior to submission of an Investigational New Drug application (IND) a sponsor can request a meeting to evaluate and reach agreement on the design of animal studies needed to initiate human testing as well as CMC and clinical aspects. Pre-IND meetings are not a requirement before submitting an IND but can be very valuable in planning a drug development program and can reduce overall time to market. The FDA’s main role at this early stage in the drug development process is to provide guidance to sponsors. Additionally, either during a Pre-IND meeting or during review of the available data included in the IND, FDA’s goal is to ensure that the compound is reasonably safe to move forward into clinical testing in the manner proposed by the clinical protocol. To obtain maximum benefit from interactions with FDA, it is important for sponsors to identify potential issues with their development program that they can address early in the program. Furthermore, discussing the clinical plan will allow FDA to assist in guiding the development pathway.
EOP1 Meetings
End-of-phase 1 (EOP1) meetings are formal PDUFA Type B meetings held between IND sponsors and CDER or CBER. Only biologics to treat serious or life- threatening illnesses (21 CFR 312 subpart E) and drugs under accelerated approval for serious/life threatening illnesses (21 CFR 314 subpart H) are eligible to request EOP1 meetings with the FDA. The goal of this meeting is to review phase 1 studies and reach agreement on plans for the phase 2 program. These meetings are especially important for sponsors wishing to submit an NDA/BLA under the accelerated approval regulations.
EOP2A Meetings
End-of-phase 2A (EOP2A) meetings are formal PDUFA Type C meetings held between IND sponsors and CDER or CBER. EOP2A meetings provide an opportunity for in-depth, exploratory discussion with the FDA focused on optimizing next steps in drug development. The overall purpose of these meetings is to discuss options for trial designs, modeling strategies, and clinical trial simulation scenarios to improve the quantification of the exposure response information from early drug development. The overall objectives of EOP2A meetings are to aid in selection of dosing regimens for the next phase of drug development and to design well planned dose-response trials that will inform later phase clinical trials by best incorporating prior quantitative knowledge.
EOP2 Meetings
End-of-phase 2 (EOP2) or Pre-phase 3 meetings are formal PDUFA Type B meetings held between IND sponsors and CDER or CBER. This type of meeting can be very useful in minimizing wasteful expenditures of time and money, speeding up the drug development and evaluation process. The purpose of an EOP2 meeting is to determine the safety of proceeding to phase 3, evaluate the phase 3 plan and protocols and the adequacy of current studies and plans to assess pediatric safety and effectiveness, and to identify any additional information necessary to support a marketing application for the uses under investigation. Although not required these meetings are critically important to the success of a development program. Sponsors who choose to utilize EOP2 Meetings have been found to have significantly higher NDA approval rates than those who do not.
Pre-NDA/Pre-BLA Meetings
Pre-New Drug Application (Pre-NDA) and Pre-Biologic License Application (Pre-BLA) meetings are formal PDUFA Type B meetings held between a sponsor and CDER or CBER, respectively. Pre-NDA/Pre-BLA meetings are critical for ensuring the submission is well-organized and readily reviewable. The purpose of a Pre-NDA/Pre-BLA meeting is to confirm that the data obtained from the registration trial(s) supports the submission, uncover any major unresolved problems, recognize studies that the sponsor is relying on as adequate and well controlled to establish the drug’s effectiveness, identify the status of ongoing or needed studies as adequate to assess pediatric safety and effectiveness, introduce FDA reviewers to the general information to be submitted, and discuss the best way to present and format data to be included in the NDA/BLA.
Summary
There are many different opportunities for sponsors to interact with FDA throughout product development. The type of interaction will depend on where a product is in the development process and the guidance being requested. Each meeting type will have its own requirements regarding meeting requests, sponsor provided documents, and timelines.
For additional information about each individual critical juncture meetings, we will talk about them in the upcoming blog posts. Please stay tuned for more!

BLOG POST BY

Theresa Downey
Regulatory Scientist, Ph.D.

Questions? Get the answer by our expert team
No two product developments are the same, talk to our experts about your development challenges and we will provide your actional recommendations.