FDA Meeting Series: How, When and What – pre-IND Meetings

Published on: Aug 16th, 2022
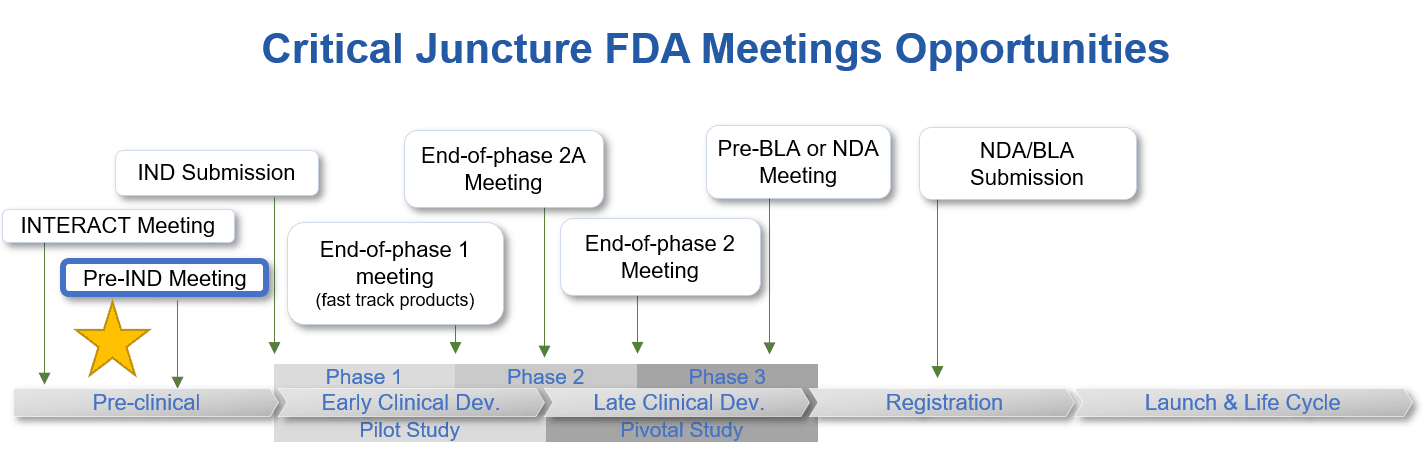
pre-IND Meetings
What:
pre-Investigational New Drug application (pre-IND) meetings are formal PDUFA Type B meetings held between a sponsor and the Center for Drug Evaluation and Research (CDER) or the Center for Biological Evaluation and Research (CBER). Prior to submission of an Investigational New Drug application (IND) a sponsor can request a meeting to evaluate and reach agreement on the design of animal studies needed to initiate human testing. Typically, only one pre-IND meeting is granted per IND. pre-IND advice may be requested for issues concerning the following:
-
Design of nonclinical pharmacology, toxicology, and drug activity studies, including design and potential uses of any proposed treatment studies in animal models
-
Data requirements, presentation, and formatting for the IND
-
Initial drug development plans, and regulatory requirements for demonstrating safety and efficacy.
-
Design of Phase I testing
-
Plans for studying the drug product in pediatric populations
-
CMC-related aspects such as unique manufacturing process, preliminary specifications, unusual excipients, viral clearance studies (for mammalian cell-derived recombinant proteins)
pre-IND meetings are not a requirement before submitting an IND but can be very valuable in planning a drug development program. These meetings can reduce time to market by:
-
Identifying and avoiding unnecessary studies
-
Ensuring that necessary studies are designed to provide useful information
-
Gaining FDA support for a proposed strategy
-
Minimizing the potential for a clinical hold
-
Providing opportunity for creative exchange of ideas
-
Obtaining regulatory insight
-
Minimizing costs
-
Clearly defining endpoints and goals of the development program
-
Allowing early interactions/negotiations with FDA
When:
A pre-IND meeting should be conducted prior to the definitive toxicology study for biological products. Typically, 1 year prior to a planned IND submission. If a biological product sponsor would like FDA input earlier in their development program an INTERACT meeting may be more appropriate. For small molecule drug products, a pre-IND meeting is typically held 6 months prior to IND submission. It is not necessary to hold this meeting prior to toxicology studies for small molecules as regulatory authorities require a standard battery of studies prior to dosing humans.
How:
If this is the first interaction with FDA a PIND number must be requested. The PIND number should be requested 30 days prior to the planned meeting request submission date from CDER (CDERAPPNUMREQUEST@fda.hhs.gov or CDER NextGen Portal) or CBER (CBERRIB@fda.hhs.gov).
pre-IND meetings must be requested by submitting a written request to the FDA via the respective center’s document room (paper submission) or via the FDA Electronic Submission Gateway (FDA ESG). Requests should be addressed to the appropriate review division or office. Meeting requests sent by fax or email are considered courtesy copies and are not a substitute for a formal written request. The meeting request should include at a minimum (additional requirements vary by reviewing division):
-
Product name
-
Chemical name, established name, and/or structure
-
Proposed indication(s) or context of product development
-
Type of meeting requested – Type B
-
Purpose of meeting/meeting objectives
-
Proposed agenda, with timeframes for each item
-
A list of proposed questions grouped by FDA discipline including a brief explanation of each question’s context and purpose
-
A list of all participants, with their titles and affiliations, who will be in attendance from the sponsor’s organization including consultants and interpreters
-
Proposed meeting dates and times including any dates and times when the sponsor is not available
-
Requested FDA attendees
-
Date meeting package will be sent
The FDA will respond to the pre-IND meeting request within 21 calendar days of request receipt. If a meeting is granted the meeting will be scheduled within 60 calendar days from the receipt of request. A briefing document of approximately 50 – 100 pages must be prepared and submitted no later than 30 days before the scheduled meeting date. The briefing document should include the same elements as the meeting request plus the following additional information:
-
Chemistry, manufacturing, and controls
-
Active Pharmaceutical Ingredient (API), Drug Product, and Placebo manufacturing methods
-
Stability, characterization, analytical methods, and release assays
-
Viral clearance (for mammalian cell line-derived recombinant proteins)
-
-
Nonclinical studies
-
In vivo and in vitro pharmacology
-
Pharmacokinetics
-
Single and multiple dose toxicity studies
-
-
Clinical development plan
-
Background and medical rationale
-
Indication to be studied
-
Natural history of the disease/condition to be treated or prevented
-
Current standard of care
-
Rationale for use of the proposed drug for the described indication
-
-
General investigation approach
-
Previous human experience (if applicable)
-
Planned clinical trials
-
If a face-to-face, teleconference or videoconference meeting is granted the FDA will provide the sponsor preliminary written responses no later than 2 days before the scheduled meeting. If the feedback is easily understood and the sponsor needs no additional clarification regarding their questions, they may cancel the meeting and the initial feedback will be sent to the sponsor as the final meeting application feedback. If the sponsor wishes to proceed with the scheduled meeting, they should choose to focus on only the questions which have not been fully resolved by the initial feedback as the meeting will only be 1 hour long. During the meeting sponsors are encouraged to listen closely, take excellent notes and be objective. The FDA will provide meeting minutes to the sponsor no later than 30 days after the meeting is held.
If a written response only (WRO) has been requested or the FDA chooses to provide WRO, the FDA will respond to the meeting request with the date they intend to send the written responses to the sponsor, generally 60 days from the meeting request receipt date. FDA will entertain requests for clarification or potential disputes to FDA responses within 1 week after receipt of the WRO. It is strongly recommended to keep this to a minimal number (1 or 2 at most). If too many questions are included FDA will recommend submission of a new meeting request for further discussion. If a meeting is denied, the FDA will notify the sponsor within 21 calendar days and will include an explanation of the reason for denial.
BLOG POST BY

Theresa Downey
Regulatory Scientist, Ph.D.

Questions? Get the answer by our expert team
No two product developments are the same, talk to our experts about your development challenges and we will provide your actional recommendations.