FDA Meeting Series: How, When and What – EOP Meetings

Published on: Aug 25th, 2022
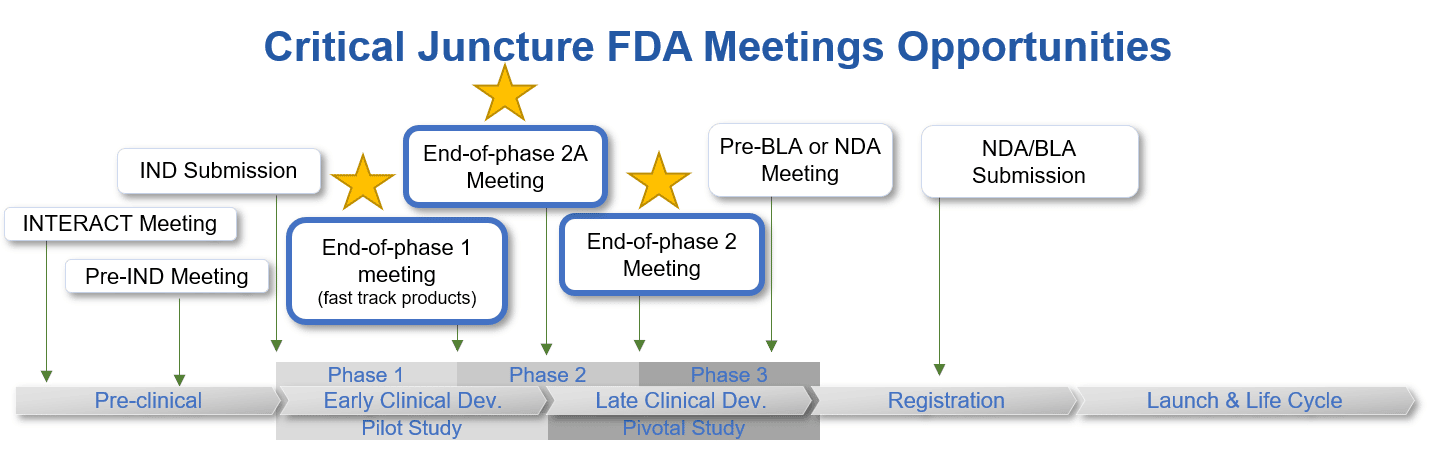
End-of-phase (EOP) meetings occur when development has reached the end of a particular phase and is ready to move to the next. There are 3 types of EOP meetings that can occur at critical junctures within a drug development program: End-of-phase 1 meeting, End-of-phase 2A meetings, and End-of-phase 2 meetings.
EOP1 Meetings
What:
End-of-phase 1 (EOP1) meetings are formal PDUFA Type B meetings held between IND sponsors and CDER or CBER. Only biologics to treat serious or life- threatening illnesses (21 CFR 312 subpart E) and drugs under accelerated approval for serious/life threatening illnesses (21 CFR 314 subpart H) are eligible to request EOP1 meetings with the FDA. The goal of this meeting is to review phase 1 studies and reach agreement on plans for the phase 2 program. Several topics may be covered in EOP1 meetings including:
-
Pharmacokinetics/Pharmacodynamics
-
Proposed phase 2 protocol
-
Identification of populations for phase 3 trials
-
Pediatric studies
-
Adaptive trial designs
-
Adequacy of supporting nonclinical data
-
Proposed studies/trials to support evaluation of abuse potential
-
Adequacy of data quality measures
-
Use of in silico methods to model PK or PD aspects of the product and use the results to eliminate or minimize the number of Phase 2 studies needed
-
Agreement on acceptability of proposed surrogate or intermediate clinal endpoints that will be the basis for the NDA/BLA
-
design of confirmatory trial(s) to demonstrate that surrogates or intermediate clinical endpoints are indicative of clinical benefit
When:
EOP1 meetings are held once phase 1 clinical data is available and prior to the initiation of phase 2 clinical studies.
How:
EOP1 meetings must be requested by submitting a written request to the FDA via the respective center’s document room (paper submission) or via the FDA Electronic Submission Gateway (FDA ESG). Requests should be addressed to the appropriate review division or office. Meeting requests sent by fax or email are considered courtesy copies and are not a substitute for a formal written request. The meeting request should include (additional requirements vary by reviewing division):
-
Application number
-
Product name
-
Chemical name, established name, and/or structure
-
Proposed regulatory pathway
-
Proposed indication(s) or context of product development
-
Type meeting being requested – Type B (EOP)
-
Pediatric study plans, if applicable
-
Human factors engineering, if applicable
-
Combination product information, if applicable
-
Proposed meeting dates and times including when the requester is not available
-
A list of proposed questions grouped by FDA discipline including a brief explanation of each question’s context and purpose
-
Proposed meeting format
-
The date the meeting background package will be sent by the sponsor
-
Purpose of meeting/meeting objectives
-
A proposed agenda, including estimated times needed for discussion such as:
-
5 minutes for introductions
-
50 minutes for discussion of questions
-
5 minutes for recap
-
-
List of sponsor attendees including consultants and interpreters
-
Requested FDA attendees
The FDA will respond to the EOP1 meeting request within 14 calendar days of request receipt. If a meeting is granted the meeting will be scheduled within 70 calendar days from the receipt of request. A meeting package must be prepared and submitted no later than 50 days before the scheduled meeting date. The meeting package should include the same elements as the meeting request plus the following additional information:
-
A background section that includes:
-
A brief history of the development program and previous relevant communications with the FDA
-
Any substantial changes in the product development plans, if applicable
-
The current status of product development
-
-
A final list of questions grouped by FDA discipline including an explanation of each question’s context and purpose
-
Data to support discussion organized by FDA discipline and question
If a face-to-face, teleconference or videoconference meeting is granted the FDA will provide the sponsor preliminary written responses no later than 5 days before the scheduled meeting. If the feedback is easily understood and the sponsor needs no additional clarification regarding their questions, they may cancel the meeting and the initial feedback will be sent to the sponsor as the final meeting minutes. If the sponsor would like to proceed with the scheduled meeting, they have the opportunity to respond to the FDA preliminary responses no later than 3 days after the receipt of preliminary responses. In practice FDA expects the responses 24 hours prior to the meeting. One example of what can be included in a response is the presentation of an alternative trial design. During the meeting sponsors are encouraged to listen closely, take excellent notes and be objective. The FDA will provide meeting minutes to the sponsor no later than 30 days after the meeting is held.
If a written response only (WRO) has been requested or the FDA chooses to provide WRO, the FDA will respond to the meeting request with the date they intend to send the written responses to the sponsor, generally 70 days from the meeting request receipt date. FDA will entertain requests for clarification or potential disputes to FDA responses within 1 week after receipt of the WRO. It is strongly recommended to keep this to a minimal number (1 or 2 at most). If too many questions are included FDA will recommend submission of a new meeting request for further discussion. If a meeting is denied, the FDA will notify the sponsor within 14 calendar days and will include an explanation of the reason for denial.
EOP2A Meetings
What:
End-of-phase 2A (EOP2A) meetings are formal PDUFA Type C meetings held between IND sponsors and CDER or CBER. EOP2A meetings provide an opportunity for in-depth, exploratory discussion with the FDA focused on optimizing next steps in drug development. The overall purpose of these meetings is to discuss options for trial designs, modeling strategies, and clinical trial simulation scenarios to improve the quantification of the exposure response information from early drug development. The overall objectives of EOP2A meetings are to aid in selection of dosing regimens for the next phase of drug development and to design well planned dose-response trials that will inform later phase clinical trials by best incorporating prior quantitative knowledge. Topics that may be discussed during EOP2A meetings include:
-
Use of quantitative information for dose selection using mechanistic or empirical relationships among biomarker, surrogate endpoints, or clinical endpoints for both effectiveness and safety
-
Use of quantitative knowledge of drug effects in animals and human subjects to aid in both dose-ranging trial design and safety assessment
-
Use of available preclinical and clinical exposure-response data and discussion of implications for dose-response trial design
-
Contrasting alternative trial design strategies and analyses
-
Use of pharmacogenetic information from preclinical studies and clinical trials and the implications of genetic factors on pharmacokinetics and/or pharmacodynamics
-
Discussion of blood or DNA sampling strategies and other trial design features to optimize the usefulness of future trials
-
Discussion of the utility of pharmacokinetic and pharmacodynamic data for dosing adjustments in special populations
When:
EOP2A meetings are held once phase 2A clinical data is available and prior to the initiation of phase 2B and phase 3 clinical studies.
How:
EOP2A meetings must be requested by submitting a written request to the appropriate Division Director within the Office of New Drugs’ document room (paper submission) or via the FDA Electronic Submission Gateway (FDA ESG). The request should specifically state that it is for an EOP2A meeting and ask that the request be forwarded immediately to the Director of Pharmacometrics in the Office of Clinical Pharmacology and the Director of the Office of Biostatistics as all three directors will work together to determine if a meeting will be granted. The following information should be included in the meeting request:
-
List of objectives, specific issues for discussion, and expected meeting outcomes
-
A list of all individuals (with titles) from the sponsor’s organization including consultants and interpreters who will attend the proposed meeting
-
The list should provide names of scientific experts (and the preferred channel of communication) who can provide clarification on the data sets and/or the quantitative analyses to facilitate communication between FDA scientific staff and the sponsor’s counterparts.
-
-
A list of completed trials describing key design features
-
Summary reports of the modeling and simulation to support the proposed trial design and dose range selection
-
Questions about drug development issues, including trial duration, dose individualization, pharmacogenomics, and data analyses
-
Proposed trial designs or analysis methods if they are to be discussed
-
Current Investigator’s Brochure and relevant literature and/or public information
Sponsors are also encouraged to submit all relevant information with the meeting request, including data, any models or simulations of trial design, or disease or outcome models that have been explored that provide insight into the issues for discussion. However, sponsors may choose to submit the electronic data and model files only if the meeting is granted. If the sponsor requests that the FDA develop model(s) for the submitted data, the sponsor should request a meeting date at least 10 weeks after FDA’s receipt of the meeting package. While FDA modeling is feasible, their resources are limited; therefore, their decisions to perform modeling work, including timelines, are made on a case-by-case basis.
The FDA will respond to the meeting request withing 21 days of request receipt. If a meeting is granted it will be scheduled within 75 days of the meeting request receipt. If any electronic data or model files were withheld, they should be shared with the FDA at this time. The FDA will provide preliminary responses no later than 5 days prior to the date of the scheduled meeting. If the feedback is easily understood and the sponsor needs no additional clarification regarding their questions, they may cancel the meeting and the initial feedback will be sent to the sponsor as the final meeting application feedback. If the sponsor would like to proceed with the scheduled meeting, they have the opportunity to respond to the FDA preliminary responses no later than 3 days after the receipt of preliminary responses. The exploratory nature of the analyses and discussions at an EOP2A meeting are intended to result in suggestions and options to assist the sponsor in optimizing the next steps of drug development. During the meeting sponsors are encouraged to listen closely, take excellent notes and be objective. The FDA will provide meeting minutes and any additional FDA-conducted modeling and simulation materials derived from sponsor-provided data to the sponsor no later than 30 days after the meeting is held.
If the FDA chooses to provide WRO the FDA will respond to the meeting request with the date they intend to send the written responses to the sponsor, generally 75 days from the meeting request receipt date. FDA will entertain requests for clarification or potential disputes to FDA responses within 1 week after receipt of the WRO. It is strongly recommended to keep this to a minimal number (1 or 2 at most). If too many questions are included FDA will recommend submission of a new meeting request for further discussion. If a meeting is not granted, the FDA will notify the sponsor within 21 calendar days of the meeting request receipt and will include an explanation of the reason for denial.
EOP2 Meetings
What:
End-of-phase 2 (EOP2) or Pre-phase 3 meetings are formal PDUFA Type B meetings held between IND sponsors and CDER or CBER. This type of meeting can be very useful in minimizing wasteful expenditures of time and money, speeding up the drug development and evaluation process. The purpose of an EOP2 meeting is to:
-
Determine the safety of proceeding to phase 3
-
Evaluate the phase 3 plan and protocols and the adequacy of current studies and plans to assess pediatric safety and effectiveness
-
Identify any additional information necessary to support a marketing application for the uses under investigation
EOP2 meetings are typically held for IND’s involving new molecular entities or drastically new uses of marketed drugs but the sponsor of any IND may request and be granted an EOP2 meeting.
When:
While the intent of EOP2 meetings is not to delay the transition of a development program from phase 2 to phase 3, they should be held before major commitments of effort and resources to specific phase 3 testing have been made to be most beneficial.
How:
EOP2 meetings must be requested by submitting a written request to the FDA via the respective center’s document room (paper submission) or via the FDA Electronic Submission Gateway (FDA ESG). Requests should be addressed to the appropriate review division or office. Meeting requests sent by fax or email are considered courtesy copies and are not a substitute for a formal written request. The meeting request should include (additional requirements vary by reviewing division):
-
Application number
-
Product name
-
Chemical name, established name, and/or structure
-
Proposed regulatory pathway
-
Proposed indication(s) or context of product development.
-
The meeting type being requested – Type B (EOP)
-
Pediatric study plans, if applicable.
-
Human factors engineering, if applicable
-
Combination product information, if applicable
-
Proposed meeting dates and times including dates when the requestor is not available
-
A list of proposed questions grouped by FDA discipline including a brief explanation of each question’s context and purpose
-
The proposed meeting format
-
The date the meeting background package will be sent by the sponsor
-
Purpose of meeting/meeting objectives
-
A proposed agenda, including estimated times needed for discussion of each topic
-
List of sponsor attendees including consultants and interpreters
-
Requested FDA attendees
The FDA will respond to the EOP2 meeting request within 14 calendar days of request receipt. If a meeting is granted the meeting will be scheduled within 70 calendar days from the receipt of request. A meeting package must be prepared and submitted no later than 50 days before the scheduled meeting date. The meeting package should include the same elements as the meeting request plus the following additional information:
-
The dosage form, route of administration, and dosing regimen (frequency and duration)
-
A background section that includes:
-
A brief history of the development program and previous relevant communications with the FDA, including a summary of all conical and nonclinical studies conducted thus far and the ones planned for NDA/BLA
-
Any substantial changes in the product development plans, if applicable
-
The current status of product development
-
-
A final list of questions grouped by FDA discipline including an explanation of each question’s context and purpose
-
Data to support discussion organized by FDA discipline and question
If a face-to-face, teleconference or videoconference meeting is granted the FDA will provide the sponsor preliminary written responses no later than 5 days before the scheduled meeting. If the feedback is easily understood and the sponsor needs no additional clarification regarding their questions, they may cancel the meeting and the initial feedback will be sent to the sponsor as the final meeting minutes. If the sponsor would like to proceed with the scheduled meeting, they have the opportunity to respond to the FDA preliminary responses no later than 3 days after the receipt of preliminary responses. During the meeting sponsors are encouraged to listen closely, take excellent notes and be objective. The FDA will provide meeting minutes to the sponsor no later than 30 days after the meeting is held.
If a written response only (WRO) has been requested or the FDA chooses to provide WRO they will respond to the meeting request with the date they intend to send the written responses to the sponsor, generally 70 days from the meeting request receipt date. FDA will entertain requests for clarification or potential disputes to FDA responses within 1 week after receipt of the WRO. It is strongly recommended to keep this to a minimal number (1 or 2 at most). If too many questions are included FDA will recommend submission of a new meeting request for further discussion. If a meeting is denied, the FDA will notify the sponsor within 14 calendar days and will include an explanation of the reason for denial.
BLOG POST BY

Theresa Downey
Regulatory Scientist, Ph.D.

Questions? Get the answer by our expert team
No two product developments are the same, talk to our experts about your development challenges and we will provide your actional recommendations.